Caso del mes febrero 2022
« Todos los casos
Caso del mes febrero 2022
Descripción
Autores
- Regina Teruel Coll
- MIR 4. [email protected]
- Joan Carreres Polo
- Radiólogo adjunto. [email protected]
- Centro de trabajo:
- Hospital Universitario y Politécnico la Fe de Valencia
Historia Clínica
Leyendas
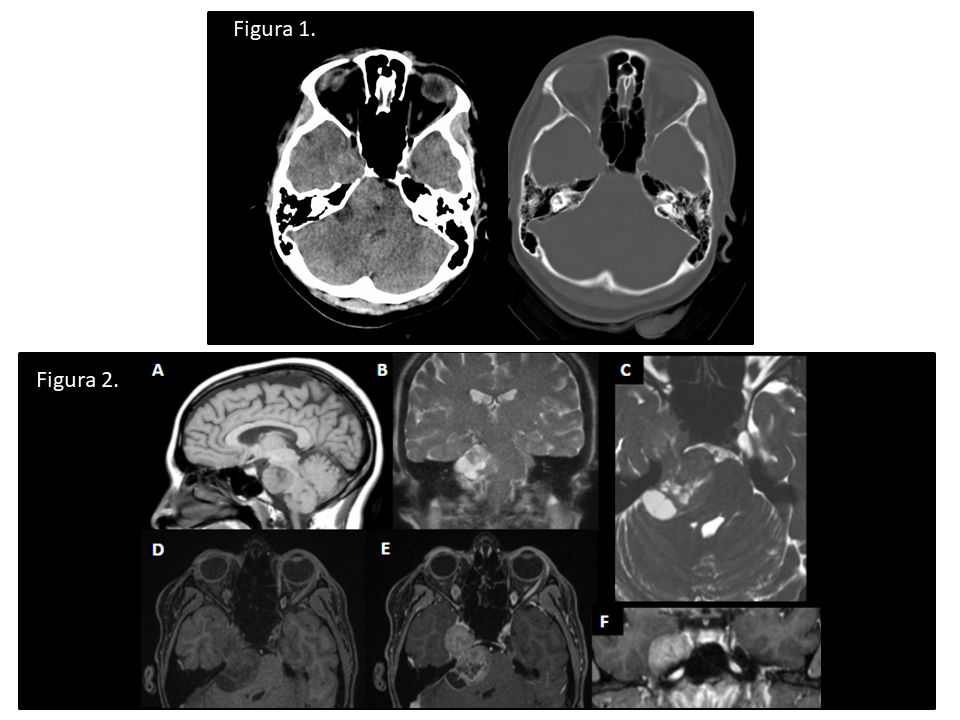
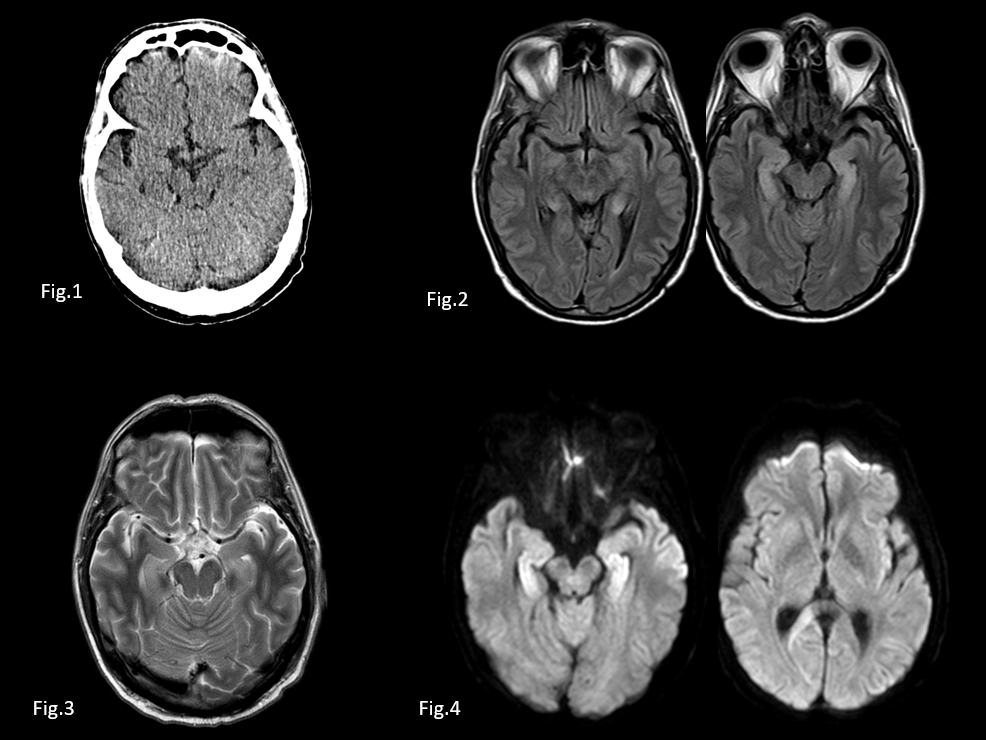
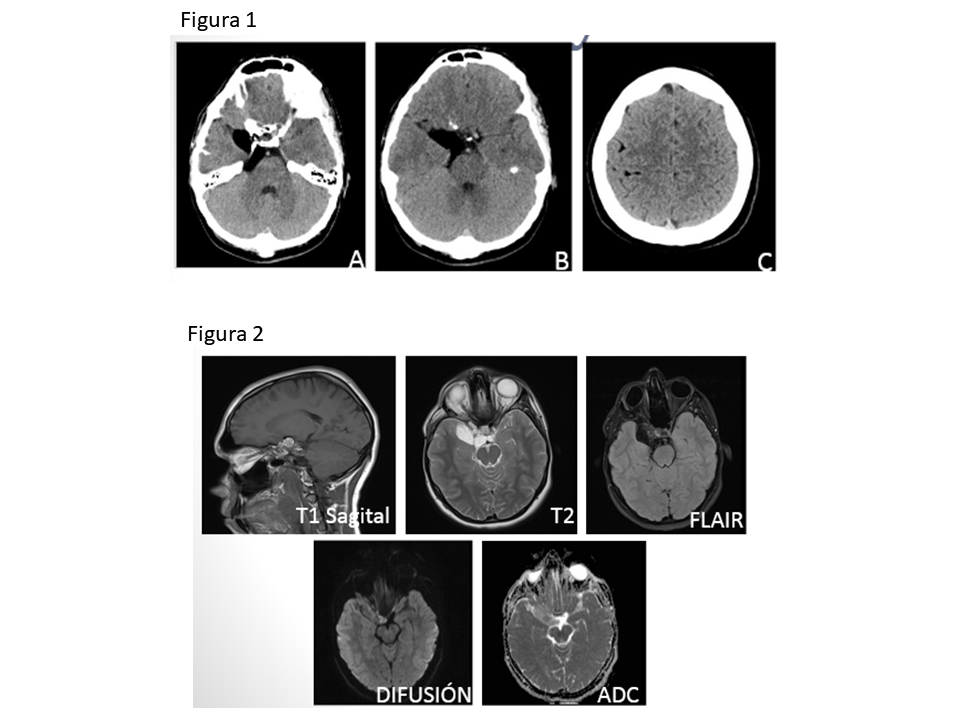
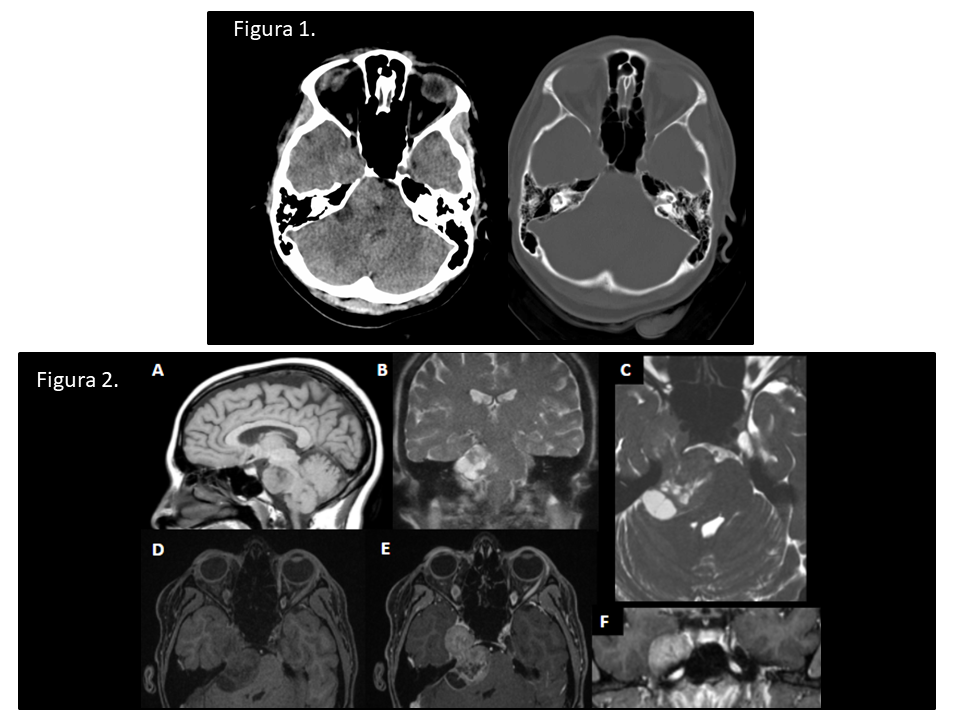
Figura 1. TC axial sin contraste IV. Masa extraaxial localizada en la cisterna prepontina con extensión paraselar derecha al cavum de Meckel. Comprime y desplaza protuberancia y deforma 4º ventrículo. Asocia remodelado óseo.
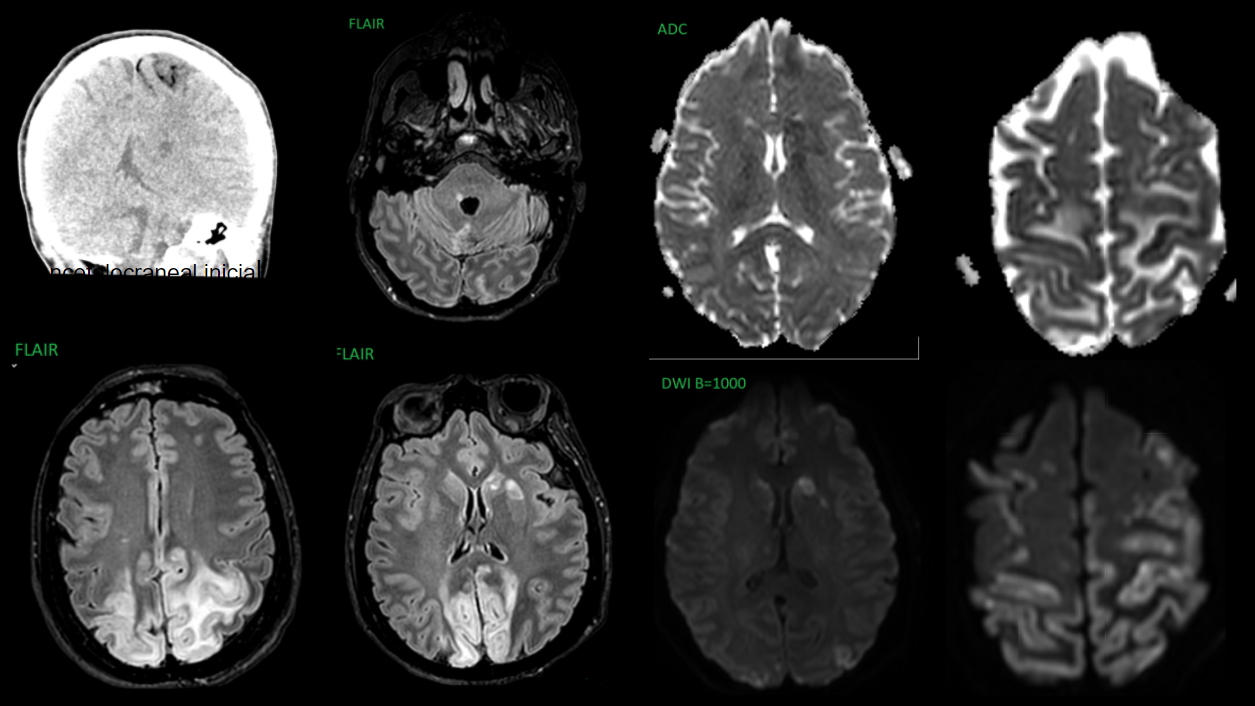
Figura 2. Imágenes de RM. Masa bilobulada en ángulo pontocerebeloso y paraselar derecha con áreas quísticas. No se identifica edema ni infiltración de estructuras adyacentes.
A. T1 axial sin contraste. Intensidad de señal intermedia.
B. T2 coronal. Componente quístico de la lesión.
C. FIESTA axial. Se identifica una hendidura de LCR que separa la vertiente posterior de la masa de la protuberancia. Vemos la constricción característica de la masa a su paso por el porus trigéminus. En el lado izquierdo se identifica el nervio trigémino contralateral sano entrando al cavum de Meckel relleno de LCR.
D y E. FAME axial pre- y postcontraste. Realce intenso tras la administración de contraste.
F. FAME coronal post-contraste. Ocupación del cavum de Meckel derecho; izquierdo de apariencia normal.
Diagnóstico
Diagnóstico.
Schwannoma del V par craneal
Los Schwannomas intracraneales son tumores benignos encapsulados de la vaina neural, característicamente de crecimiento lento. Se presentan como masas extraaxiales bien circunscritas con desplazamiento de estructuras adyacentes, pero sin invasión tisular.
En TC es característica una atenuación baja o intermedia con frecuentes cambios quísticos, así como la remodelación ósea o la expansión foraminal. Las calcificaciones y la hemorragia son poco frecuentes.
En RM, presentan isointensidad de señal respecto al córtex en T1 y señal heterogéneamente hiperintensa en T2, con un un realce intenso tras la administración de contraste.
Los Schwannomas del V par tienen suelen originarse en la unión del ganglio de Gasser con la raíz nerviosa. Las lesiones pequeñas pueden estar confinadas al Cavum de Meckel, pudiendo dar el signo del “winking Meckel Cave” consistente en el contraste entre el cavum ocupado por tumor y el contralateral sano relleno de líquido cefalorraquídeo.
Los tumores bicompartimentales, como en el caso expuesto, se originan en el cavum de Meckel y se extienden a la fosa posterior a través del porus trigeminus, con constricción a su paso por este punto, dándole la apariencia característica en mancuerna. Los tricompartimentales se producen cuando el tumor bicompartimental afecta a la rama V3 y se extiende anteroinferiormente desde la fosa craneal media a través del foramen oval al espacio masticador.
El diagnóstico diferencial cuando se presentan como tumores bi- o tricompartimentales es bastante limitado, ya que los hallazgos son característicos de esta entidad. Cuando se afecta aisladamente el cavum de Meckel, el diagnóstico diferencial se realiza principalmente con el meningioma y las metástasis, incluyendo la diseminación de tumores de cabeza y cuello. Otras lesiones con afectación del cavum de Meckel son lesiones vasculares o adenomas pituitarios.
El tratamiento varía en función de sintomatología, localización o tamaño. Pueden realizarse controles periódicos o tratamiento quirúrgico o mediante radiocirugía. En nuestro caso se realizó cirugía y se confirmó histopatológicamente el diagnóstico de Schwannoma grado 1 de la OMS.
BIBLIOGRAFÍA:
Osborn, A., Hedlund, G. & Salzman, K. (2018). Osborn’s brain : imaging, pathology, and anatomy. Philadelphia, PA: Elsevier. Barkhof, F., Jäger, R., Thurnher, M. & Rovira, A. (2019). Clinical neuroradiology : the ESNR textbook. Cham, Switzerland: Springer.