Caso del mes Febrero 2017
« Todos los casos
Caso del mes Febrero 2017
Descripción
Autores
- Dr. Alberto Vargas Solano. Sección de Neurorradiología. Servicio de Radiodiagnóstico. Hospital Universitari Clinic de Barcelona. Email: [email protected]
- Sebastián Capurro Ferrer. Sección de Neurorradiología. Servicio de Radiodiagnóstico. Hospital Universitari Clinic de Barcelona. E-mail: [email protected]
Historia Clínica
- Paciente mujer de 17 años con antecedente de incontinencia pigmentaria, migraña, síndrome depresivo, trastorno de la conducta alimentaria y síndromes sincopales.
- Actualmente valorada por cuadro depresivo y sincopal asociado a parestesias, aumento de reflejos osteotendinosos, mioclonias y debilidad de miembros inferiores, en tratamiento con carbamazepina con disminución súbita de la dosis debido a intoxicación.
Leyendas
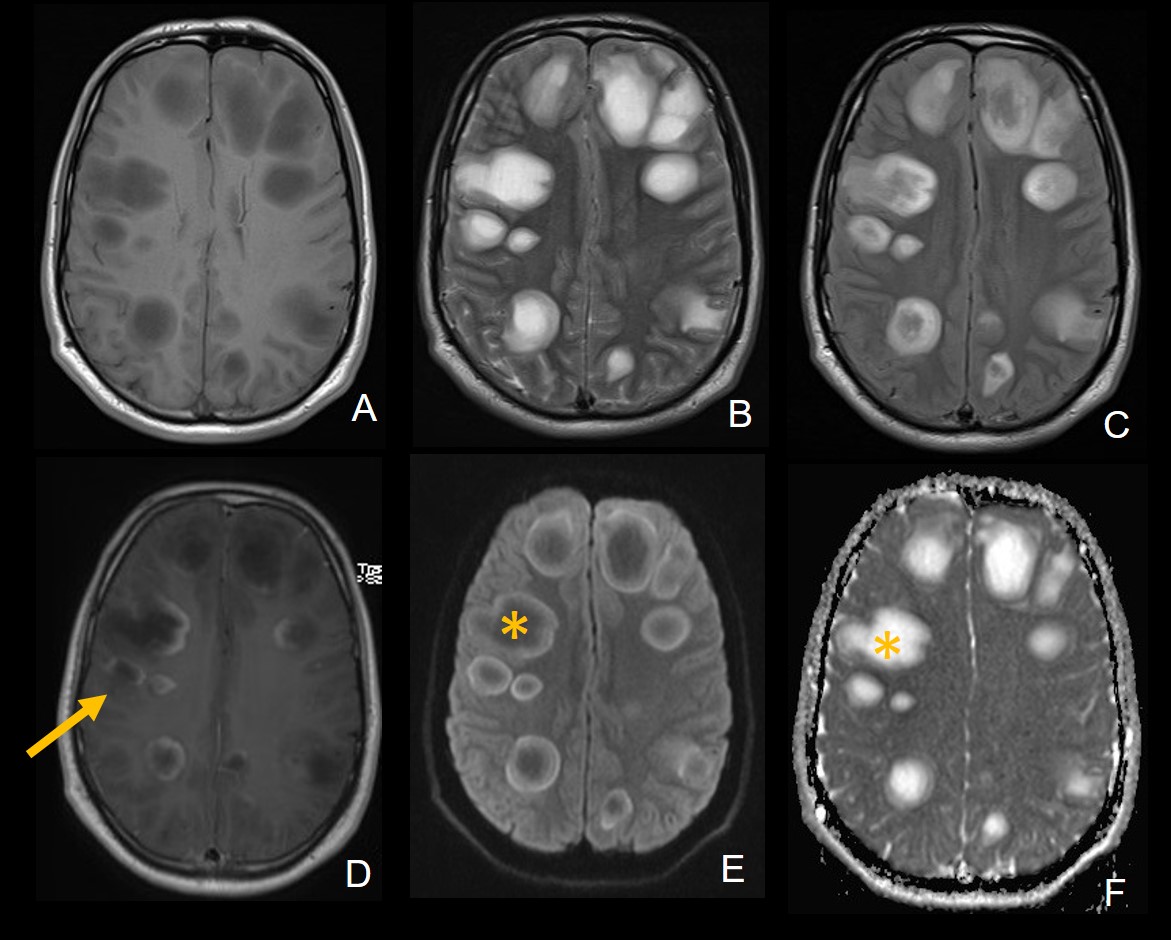
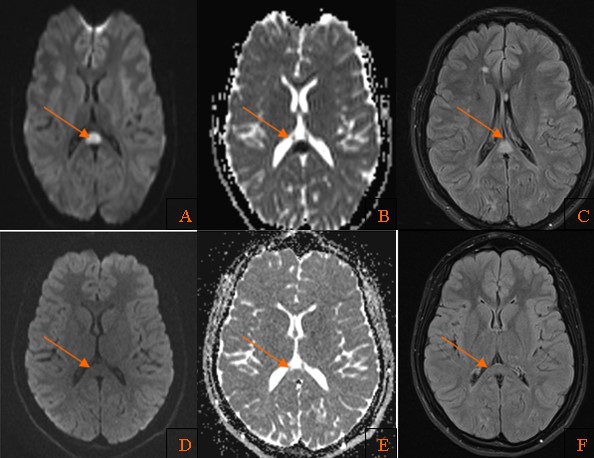
A,B,C: Imágenes axiales DWI, ADC y FLAIR que muestran imagen nodular hiperintensa en el esplenium del cuerpo calloso con restricción a la difusión.
D,E,F: Imágenes axiales DWI, ADC y FLAIR, de control un mes después de adquirido el estudio previo, con ausencia de la imagen previo mencionada o cambios glioticos residuales en el cuerpo calloso.
Diagnóstico
Diagnóstico:
Lesión evanescente del esplenium del cuerpo calloso por retirada del tratamiento antiepiléptico.
Comentario:
- Las lesiones nodulares evanescentes del esplenium del cuerpo calloso son secundarias al retiro abrupto de tratamientos antiepilépticos, sincopes, alteraciones hidroelectrolíticas, esclerosis múltiple, encefalomielitis aguda diseminada (ADEM), encefalopatía posterior reversible (PRES) y enfermedades infecciosas como el SIDA.
- Hay dos formas de presentación: focal/nodular y difusa.
- La forma nodular es característica del retiro abrupto de medicamentos antiepilépticos.
- Las demás enfermedades producen lesiones que comprometen difusamente el esplenium.
- La RM muestra una imagen nodular bien definida, hiperintensa en T2/FLAIR hipointensa en T1, con restricción en DWI/ADC, la cual desparece en controles posteriores a más tardar un mes.
Bibliografía:
- J. Takanashi. Widening Spectrum of a Reversible Splenial Lesion with Transiently Reduced Diffusion. AJNR (2006) 27, 836-838.T Polster.
- Transient lesion in the splenium of the corpus callosum: three further cases in epileptic patients and a pathophysiological hipótesis. J. Neurol Neurosurg Psychiatry 2001;70:459–463.
- Kerstin Anneken. Transient lesion in the splenium related to antiepileptic drug: Case report and new pathophysiological insights. Seizure (2008) 17, 654—657.