Caso del mes Agosto 2019
« Todos los casos
Caso del mes Agosto 2019
Descripción
Autores
- Beatriz Brea Álvarez
- Inés Hernández Delgado
- María Luisa Collado Torres
- Yumara Malo Rubio
- María Ibnoulkhatib
- Hospital Universitario Puerta de Hierro- Majadahonda
Historia Clínica
Consulta por clínica de mareos con giro de objetos que empezó hace 2 semanas coincidiendo con cuadro de gastroenteritis. La sensación de giro de objetos duro 24 horas aproximadamente y desde entonces presenta inestabilidad que se acentúa con los movimientos cefálicos. Desde hace 4 días se asocia otalgia derecha sin supuración por lo que acude a su medico de primaria quien indica tratamiento con Cetraxal que ha cumplido hasta hoy sin mejoría.
Otoscopia: OI Normal OD: CAE ocupado por otorrea con hifas negras, se observa paredes del CAE muy inflamadas. En piel de la concha se observan 3 costras hemáticas.
Juicio Diagnóstico: Otitis externa derecha en paciente imnusuprimido. Se solicita TC y posteriormente se amplía estudio con RM.
Leyendas
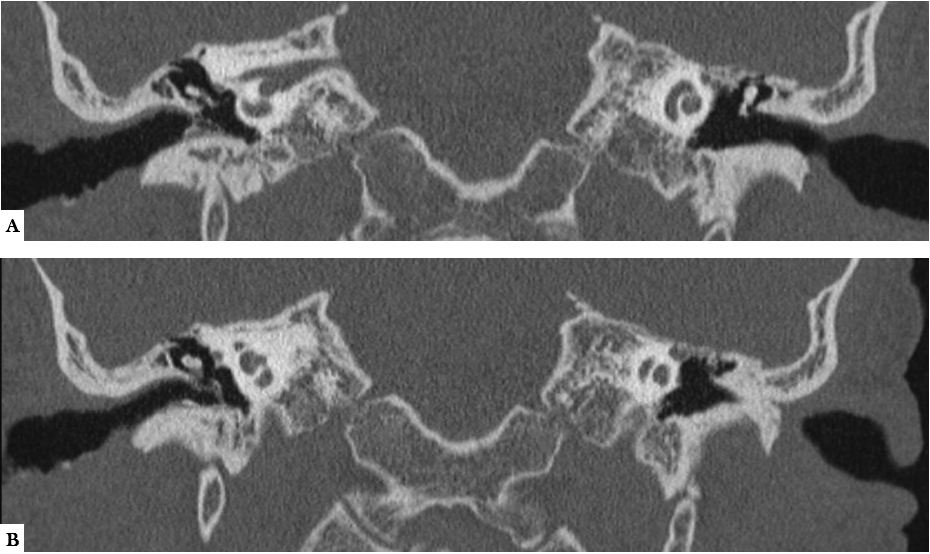
A, B. TC MPR coronal. Engrosamiento leve e irregular de las paredes del CAE y engrosamiento de la membrana timpánica derechas. No ocupación de la caja derecha (existía un discreto aumento de partes blandas en celdillas mastoideas- no mostrado).Oído izquierdo sin alteraciones.
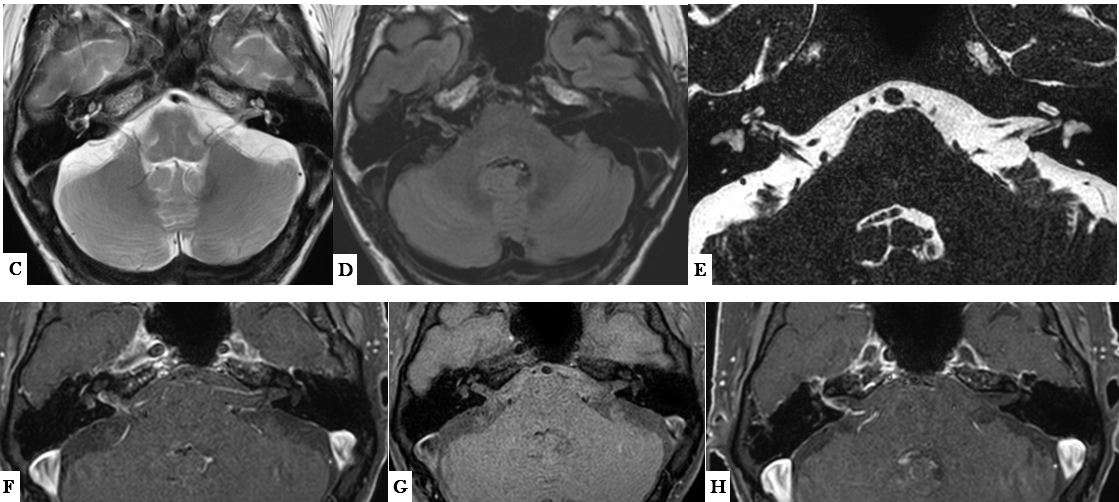
C. RM corte axial potenciado en T2. No alteraciones en la señal del tronco del encéfalo ni en el cerebelo. Ocupación discreta de la caja timpánica
D. RM corte axial portenciado en Flair. Hiperseñal en el conducto auditivo interno (CAI) y en el oído interno izquierdos con señal normal del lado izquierdo.
E. RM estudio 3DT2 –drive- con reconstrucción axial. Se identifican los nervios coclear y vestibular inferior en el CAI derecho, pero además existe un bucle vascular (se confirma rama de la AICA en el estudio angiográfico no mostrado) y una “estructura pseudolineal” que no permite definir bien el nervio vestibular.
F, G y H. RM cortes axiales T1 con saturación grasa, F y H tras administrar gadolinio. En el estudio sin gadolinio (G) existe una tenue hiperseñal de la cóclea y laberinto membranoso de forma comparativa con el lado izquierdo. Tras administrar gadolinio (F y H) existe refuerzo irregular de las paredes del CAI, segmento laberíntico y primera vuelta del nervio facial, y del vestíbulo derechos. Posible refuerzo asimétrico de la meninge derecha adyacente al borde posterior de la pirámide petrosa derecha.
Diagnóstico
Diagnóstico.
Laberintitis meningocócica (confirmada por punción lumbar).
La laberintitis es la inflamación del oído interno cuyo origen es viral en la mayoría de las ocasiones. Mas rara vez tiene un origen bacteriano por una infección meníngea.
La clínica es un cuadro vestibular asociado en la mayoría de las ocasiones a hipoacusia neurosensorial.
Se clasifica en:
– Timpanogénica. El origen es una otitis media cuyo agente patógeno accede al oído interno erosionando el laberinto óseo o a través de las ventana oval o redonda.
– Hematógena. El agente etiológico generalmente es vírico, como sucede en le caso de las paperas o el sarampión. Menos frecuente es la sífilis.
– Postraumática
– Meningogénica. La infección meníngea vírica o, más frecuente, bacteriana alcanza el oído interno a través del CAI. Es más frecuente en niños y, debido a su origen, suele ser bilateral.Radiológicamente se han descrito tres fases en la infección. En la fase aguda no existen alteraciones en la TC, existiendo únicamente un refuerzo del laberinto membranoso en la RM con gadolinio. En la fase subaguda la TC sigue siendo normal y en la RM aparece hiperseñal del laberinto en Flair y, menos significativa, en el T1 secundario a el contenido proteináceo. Además el realce con gadolinio es más evidente. La fase crónica se caracteriza por osificación del laberinto y, por lo tanto, se identifica un oído interno hiperdenso en la TC y no visualización del laberinto membranoso en la RM.
El diagnóstico diferencial radiológico es variable dependiendo de la fase de la enfermedad. En la fase aguda y ante los hallazgos clínicos, la presencia de realce es casi diagnóstico de esta entidad. En la fase subaguda la hiperseñal en Flair y, tenue hiperseñal en T1 puede plantear el diagnóstico diferencial con la hemorragia laberíntica. La presentación clínica de esta entidad suele ser como hipoacusia súbita refiriendo la mayoría a los pacientes toma de anticoagulantes u otras causas predisponentes de hemorragia. En la fase crónica la esclerosis de la pirámide petrosa- ausencia de visualización del laberinto óseo en la TC, puede plantear el diagnóstico diferencial con la aplasia del laberinto. En esta situación la pirámide suele ser más estrecha al no tener que alojar el oído interno en su interior.
BIBLIOGRAFÍA:
Abele TA. Imaging of the temporal bone. Radiol Clin North Am, 2015; 53:15-36
Juliano AF. Imaging review of the temporal bone: part I. anatomy and inflammatory and neoplastic processes. Radiology, 2013; 269: 17-33.
Lingam RK. Inflammation of the temporal Bone. Neuroimaging Clin North Am, 2019; 29: 1-17