Diagnóstico:
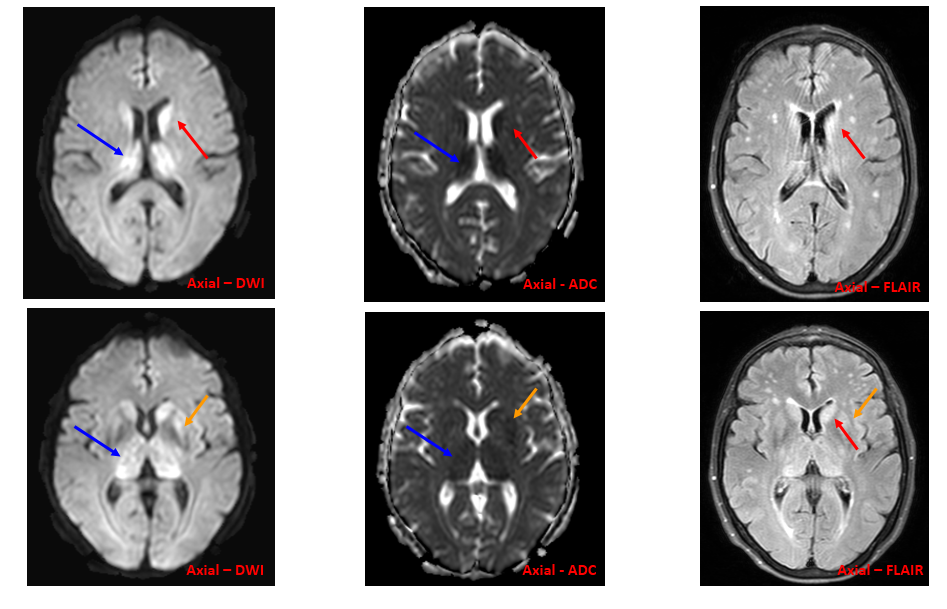
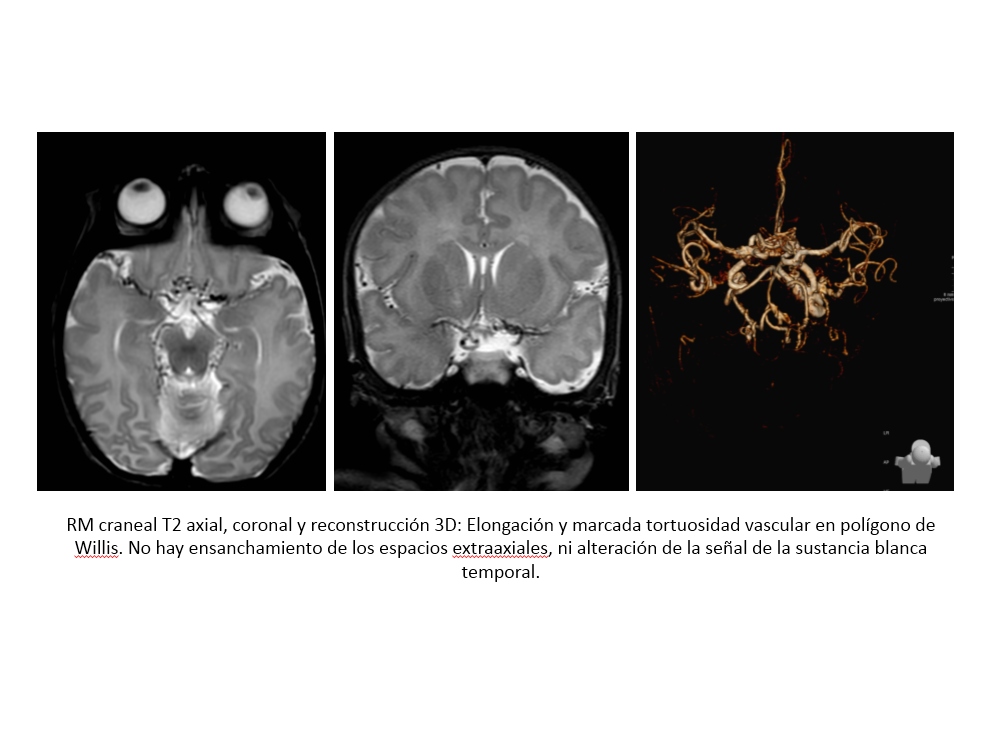
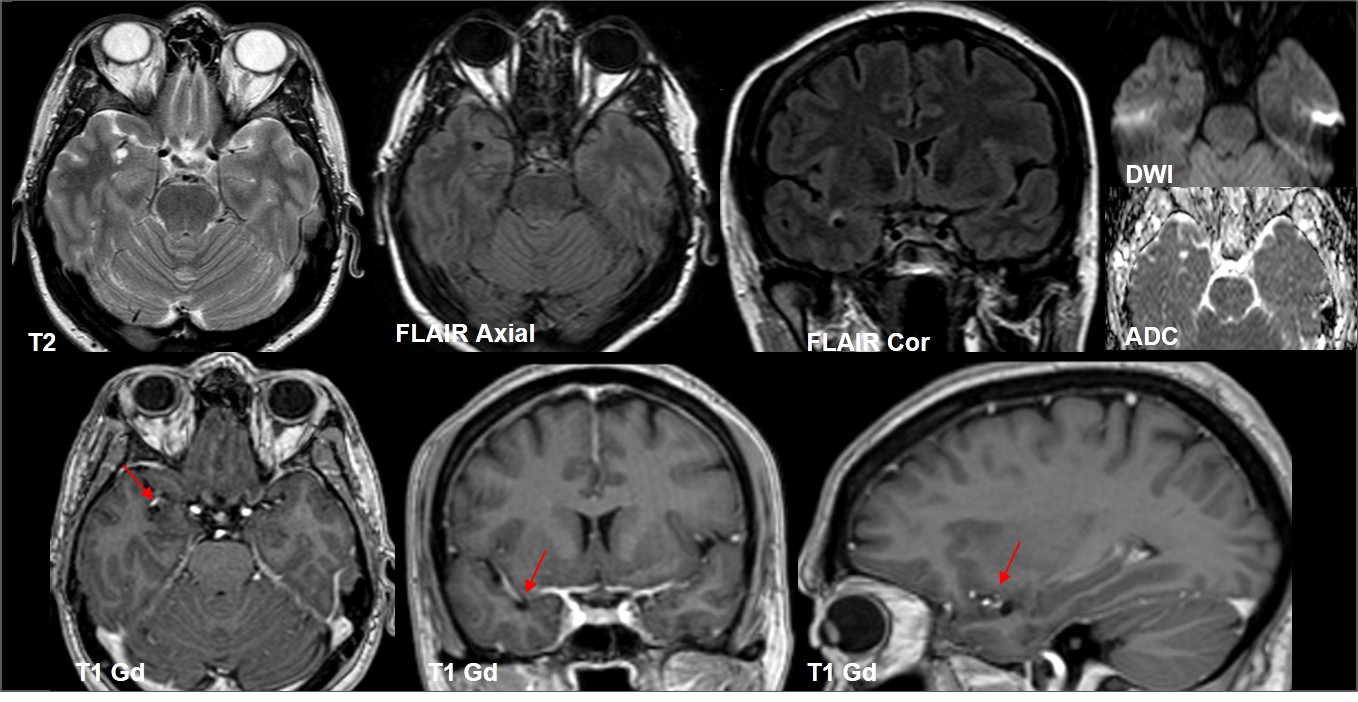
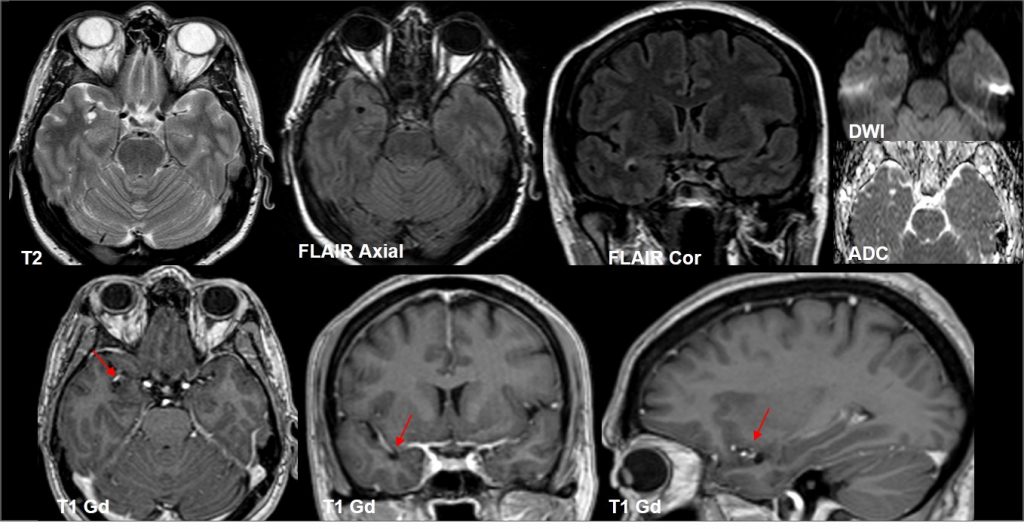
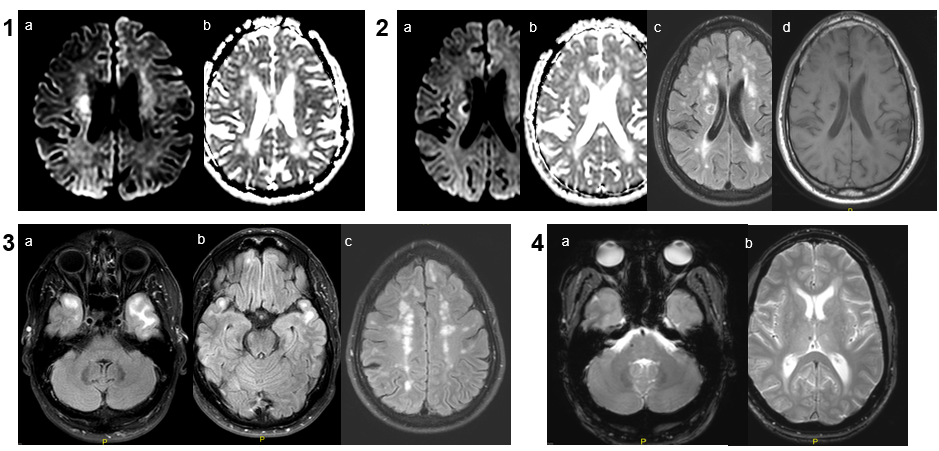
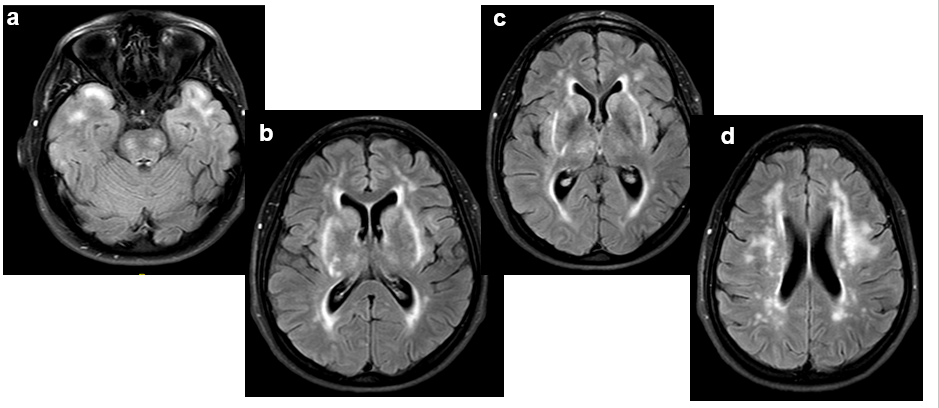
Espacio perivascular de Virchow Robin temporal anterior dilatado.
Los espacios perivasculares de Virchow Robin son espacios ocupados por líquido intersticial y rodeados de pia, que envuelven los vasos penetrantes en su curso desde el espacio subaracnoideo hacia el parénquima cerebral.
Junto con las características típicas de neuroimagen, el lugar anatómico es un criterio importante para el diagnóstico de los espacios de Virchow Robin. En los últimos años se ha descrito y publicado una variante o nueva localización de estos espacio, una localización atípica a nivel subcortical en los polos anteriores de los lóbulos temporales.
Las características de imagen típicas son:
Lesiones de entre 5 y 15 mm, redondeadas u ovaladas, de señal similar al LCR (hipointensa en T1 e hiperintensa en T2), con estabilidad de su tamaño a través del tiempo. Únicamente se han descrito 2 casos en que la lesión cambió de tamaño, por aumento en uno y por disminución en el otro.
Suelen asociar grado variable de alteración de la señal o edema perilesional en secuencias potenciadas en T2 (~80%), que ocasionalmente puede variar en el tiempo
Suelen estar contacto o próximos con una estructura vascular, que suele ser una rama de la arteria cerebral media.
Asocian zonas de adelgazamiento o distorsión de la cortical del parénquima adyacente en hasta un 90%
No realzan con el contraste.
Pueden asociar pequeños espacios perivasculares adyacentes.
Localización próxima al espacio subaracnoideo.
En el rango de diagnósticos diferenciales se incluyen neoplasias quísticas (glioma de bajo grado, ganglioglioma, tumor neuroepitelial disembrioplásico, gangliocitoma), neurocisticercosis, quiste neuroepitelial, entre otros; sin embargo los hallazgos por imagen descritos de estos espacios de VR dilatados temporales son muy característicos, logrando una gran aproximación diagnóstica en la mayoría de los casos.
BIBLIOGRAFÍA:
Lim AT, Chandra RV, Trost NM et-al. Large anterior temporal Virchow-Robin spaces: unique MR imaging features. Neuroradiology. 2015;57 (5): 491-9. doi:10.1007/s00234-015-1491-y
Rawal S, Croul SE, Willinsky RA et-al. Subcortical cystic lesions within the anterior superior temporal gyrus: a newly recognized characteristic location for dilated perivascular spaces. AJNR Am J Neuroradiol. 2014;35 (2): 317-22. doi:10.3174/ajnr.A3669
Kwee RM, Kwee TC. Virchow-Robin spaces at MR imaging. Radiographics. 2007;27 (4): 1071-86. doi:10.1148/rg.274065722