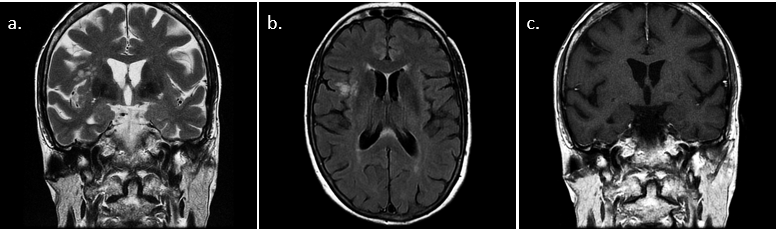
Diagnóstico.
Tumor neuronal multinodular y vacuolizante
Los tumores neuronales multinodulares y vacuolizantes (MNVT, en inglés) se han incluido recientemente en la WHO Classification of Tumors of The Central Nervous System (2016) como tumores de bajo grado derivados de células gliales y/o neuronales. Aún así, se plantea que podrían tener una naturaleza malformativa debido a la ausencia de crecimiento expansivo e infiltrativo y a su baja actividad mitótica.
En resonancia magnética se presentan como múltiples pequeños nódulos agrupados, situados supratentoriales y superficiales en la sustancia blanca subcortical, que no asocian edema vasogénico ni efecto de masa sobre estructuras adyacentes. Son hiperintensos en T2 y FLAIR, sin ser visibles en otras secuencias ni tras la administración de contraste.
La mayoría de los MNVT son asintomáticos y se diagnostican de forma incidental en pruebas de imagen, aunque algunos se presentan con clínica de cefalea o de crisis comicial. A menos que sean sintomáticos, no suelen requerir biopsia, resección o seguimiento mediante pruebas de imagen.
BIBLIOGRAFÍA:
Thom M, Liu J, Bongaarts A, Reinten R, Paradiso B, Jäger H et al. Multinodular and vacuolating neuronal tumors in epilepsy: dysplasia or neoplasia?. Brain Pathology. 2017;28(2):155-171.
Alsufayan R, Alcaide-Leon P, de Tilly L, Mandell D, Krings T. Natural history of lesions with the MR imaging appearance of multinodular and vacuolating neuronal tumor. Neuroradiology. 2017;59(9):873-883.
Nunes R, Hsu C, da Rocha A, do Amaral L, Godoy L, Watkins T et al. Multinodular and Vacuolating Neuronal Tumor of the Cerebrum: A New “Leave Me Alone” Lesion with a Characteristic Imaging Pattern. American Journal of Neuroradiology. 2017;38(10):1899-1904.